


It is a rare but serious bleeding disorder that affects the body’s ability to control bleeding. It is primarily a genetic condition, and individuals with hemophilia have a deficiency or absence of certain clotting factors, which are essential for the normal clotting process. This condition can lead to prolonged and potentially life-threatening bleeding episodes, even from minor injuries or internal bleeding. In this article, we will delve into the risk factors, causes, types, diagnosis, and medical treatment of hemophilia.
Risk Factors of Hemophilia
Hemophilia is a genetic disorder, so the most significant risk factor is having a family history of the condition. It is primarily passed down through generations due to mutations in specific genes responsible for producing clotting factors in the blood. However, there are other risk factors and considerations that can influence the likelihood of developing hemophilia or its severity:
1. Family History: As mentioned earlier, having a family history of hemophilia is the most critical risk factor. If a close family member, such as a father, brother, or maternal uncle, has hemophilia, the chances of inheriting the genetic mutation increase.
2. Gender: Hemophilia is an X-linked genetic disorder, meaning it primarily affects males. Since males have one X and one Y chromosome, if their X chromosome carries the mutated gene responsible for hemophilia, they will have the condition. Females have two X chromosomes and are typically carriers; they can pass the mutated gene to their offspring, but they themselves may not experience severe symptoms.
3. Spontaneous Mutations: In some cases, hemophilia can occur without a family history of the condition. Spontaneous mutations in the genes responsible for producing clotting factors can lead to hemophilia in individuals with no previous family history of the disorder.
4. Advanced Maternal Age: There is some evidence to suggest that the risk of spontaneous mutations leading to hemophilia may be higher in pregnancies where the mother is of advanced maternal age.
5. Consanguinity: Hemophilia is more likely to occur in families with a history of consanguineous (blood-related) marriages. The closer the biological relationship between the parents, the higher the chance of inheriting the mutated gene.
6. Hemophilia Carrier Status in Females: As mentioned earlier, females can be carriers of the hemophilia gene without experiencing significant symptoms themselves. If a woman is a carrier, she has a 50% chance of passing the mutated gene to her offspring, regardless of the child’s gender.
7. Genetic Testing: Families with a history of hemophilia may undergo genetic testing to identify carriers and assess the risk of having affected offspring.
8. Acquired Hemophilia: Although rare, acquired hemophilia can affect individuals without a family history of the condition. It occurs when the immune system produces antibodies that attack clotting factors, leading to hemophilia-like symptoms.
Causes of Hemophilia
Hemophilia is primarily caused by genetic mutations that affect the production or function of specific clotting factors in the blood. These clotting factors are essential for the normal coagulation process, which stops bleeding by forming blood clots. The two most common types of hemophilia are Hemophilia A and Hemophilia B, which are caused by deficiencies in different clotting factors:
Inheritance:
As mentioned earlier, hemophilia is an inherited disorder, and the majority of cases are passed down through generations. When a mother carries a mutated gene responsible for hemophilia on one of her X chromosomes, she is considered a carrier. If she has a son, there is a 50% chance that the son will inherit the mutated gene and develop hemophilia. Daughters of carriers have a 50% chance of becoming carriers themselves.
Spontaneous Mutations:
In some cases, hemophilia can occur without a family history of the disorder. Spontaneous mutations can lead to the development of hemophilia in individuals with no family history of the condition. These mutations can occur in the affected individual’s genes during the early stages of development.
Rare Acquired Hemophilia:
While the majority of hemophilia cases are genetic, there is a rare form known as acquired hemophilia. Acquired hemophilia is not inherited but occurs when the immune system produces antibodies that attack and inhibit clotting factors, most commonly factor VIII. This form of hemophilia can affect both males and females and can be associated with various underlying conditions, such as autoimmune diseases, cancer, or pregnancy.
Types of Hemophilia
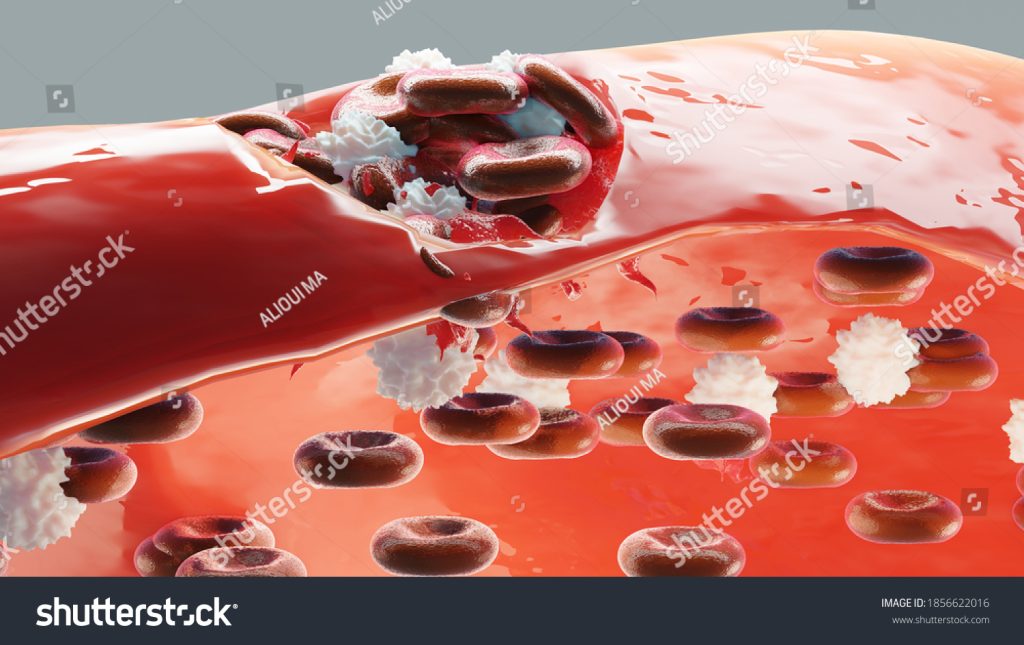
Hemophilia is classified into different types based on the deficient clotting factor present in the blood. The two most common types are Hemophilia A and Hemophilia B, but there are also other, rarer forms of the disorder. Let’s explore the types of hemophilia:
Hemophilia A (Factor VIII Deficiency):
Hemophilia A is the most common type of hemophilia, accounting for approximately 80-85% of all cases. It is caused by a deficiency or dysfunction of clotting factor VIII (FVIII). Factor VIII plays a crucial role in the coagulation cascade, helping to stabilize the formation of a fibrin clot. Hemophilia A is an X-linked disorder, which means it primarily affects males, while females are typically carriers of the mutated gene.
2. Hemophilia B (Factor IX Deficiency):
Also known as Christmas disease, Hemophilia B is the second most common type of hemophilia, accounting for about 15-20% of cases. It is caused by a deficiency or dysfunction of clotting factor IX (FIX). Factor IX is another essential component of the coagulation cascade, working together with factor VIII to ensure proper blood clotting. Like Hemophilia A, Hemophilia B is an X-linked disorder that primarily affects males, while females can be carriers of the defective gene.
3. Hemophilia C (Factor XI Deficiency):
Hemophilia C is a rarer form of hemophilia compared to Hemophilia A and B. It is caused by a deficiency of clotting factor XI (FXI). Hemophilia C is an autosomal recessive disorder, which means both parents must carry the mutated gene and pass it on to their child for the condition to develop. Unlike Hemophilia A and B, Hemophilia C affects both males and females equally.
4. Hemophilia D (Factor XII Deficiency):
Hemophilia D is an extremely rare form of hemophilia caused by a deficiency of clotting factor XII (FXII). The symptoms of Hemophilia D are generally milder compared to other types of hemophilia, and the bleeding episodes are less frequent.
5. Hemophilia E (Factor V Deficiency):
Hemophilia E is an exceedingly rare form of hemophilia caused by a deficiency of clotting factor V (FV). It is also known as Owren’s disease. Hemophilia E is typically characterized by mild to moderate bleeding symptoms.
6. Hemophilia with Inhibitors:
Some individuals with hemophilia, particularly those receiving clotting factor replacement therapy, may develop inhibitors, which are antibodies that neutralize the effects of the administered clotting factor. Hemophilia with inhibitors can complicate treatment and management, requiring specialized care.
It’s important to note that Hemophilia A and Hemophilia B are the most prevalent types, while the other forms of hemophilia are relatively rare.
Diagnosis of Hemophilia
Diagnosing hemophilia involves several steps, including a detailed medical history, physical examination, and specialized laboratory tests. The following are some essential components of the diagnosis process:
a) Medical History: The doctor will inquire about any family history of bleeding disorders, as hemophilia is often inherited. They will also ask about any past bleeding episodes, including their frequency, severity, and response to treatments.
b) Physical Examination: The doctor will perform a thorough physical examination to look for signs of bleeding, such as bruising, joint swelling, or prolonged bleeding from cuts.
c) Coagulation Tests: The definitive diagnosis of hemophilia is made by measuring the clotting factor levels in the blood. These tests, such as the activated partial thromboplastin time (aPTT) and factor assays, can determine the type and severity of hemophilia.
Medical Treatment
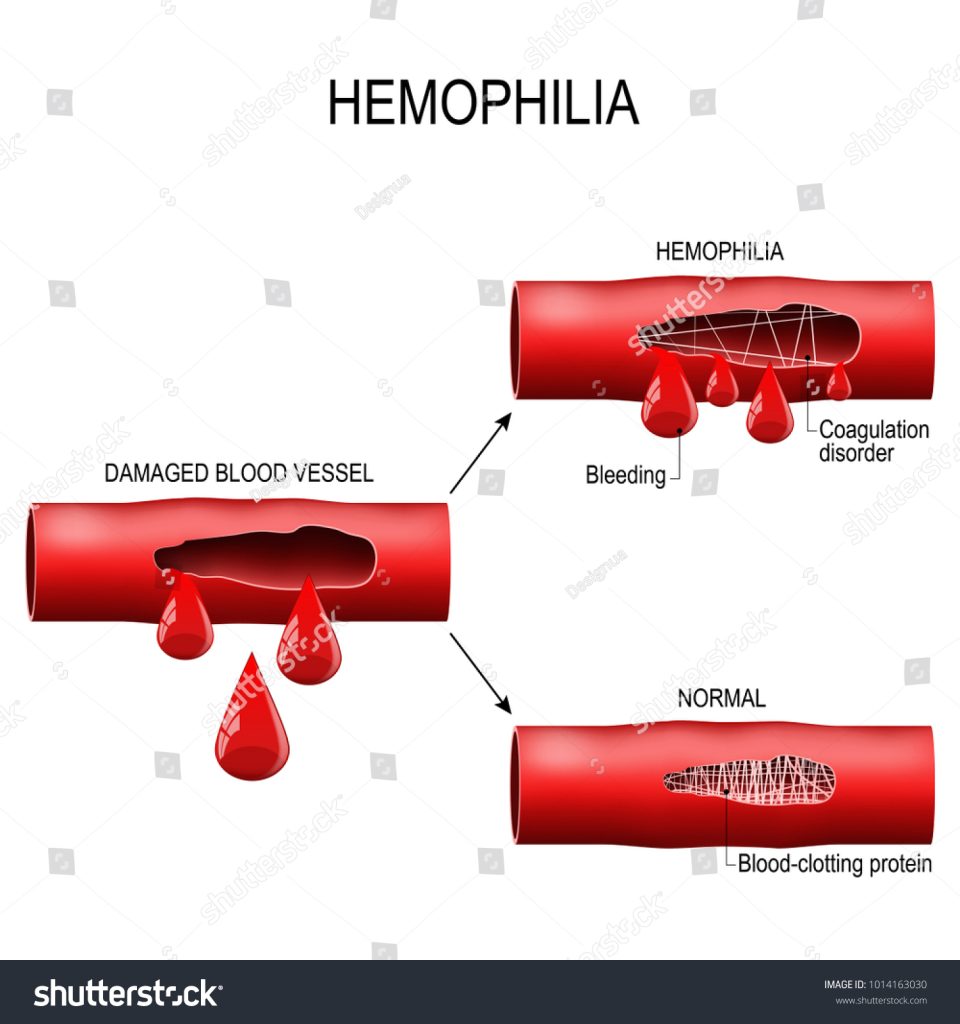
While there is currently no cure for hemophilia, several medical treatments can help manage the condition and prevent complications. The mainstay of treatment is to replace the missing clotting factor through regular infusions. This treatment is known as clotting factor replacement therapy and can be administered on-demand or as prophylaxis to prevent bleeding episodes.
a) Clotting Factor Replacement Therapy: During this treatment, the missing clotting factor (factor VIII for hemophilia A or factor IX for hemophilia B) is infused directly into the bloodstream. The therapy helps restore the blood’s ability to clot properly, reducing the risk of bleeding.
b) Desmopressin (DDAVP): In some cases of mild hemophilia A, desmopressin, a synthetic hormone, can stimulate the release of stored clotting factor VIII from the body’s tissues, temporarily raising clotting factor levels.
c) Hemostatic Agents: Topical agents, such as fibrin sealants or gelatin sponges, can be used to promote clotting and stop bleeding from minor cuts or wounds.
d) Gene Therapy: Emerging treatments involve gene therapy, where healthy copies of the defective gene are introduced into the body to produce the missing clotting factor.
e) Management of Bleeding Episodes: In the event of a bleeding episode, prompt treatment is essential to control bleeding and prevent complications. RICE (Rest, Ice, Compression, Elevation) can be used for joint bleeding, and pain management may also be necessary.
Preventive Measures
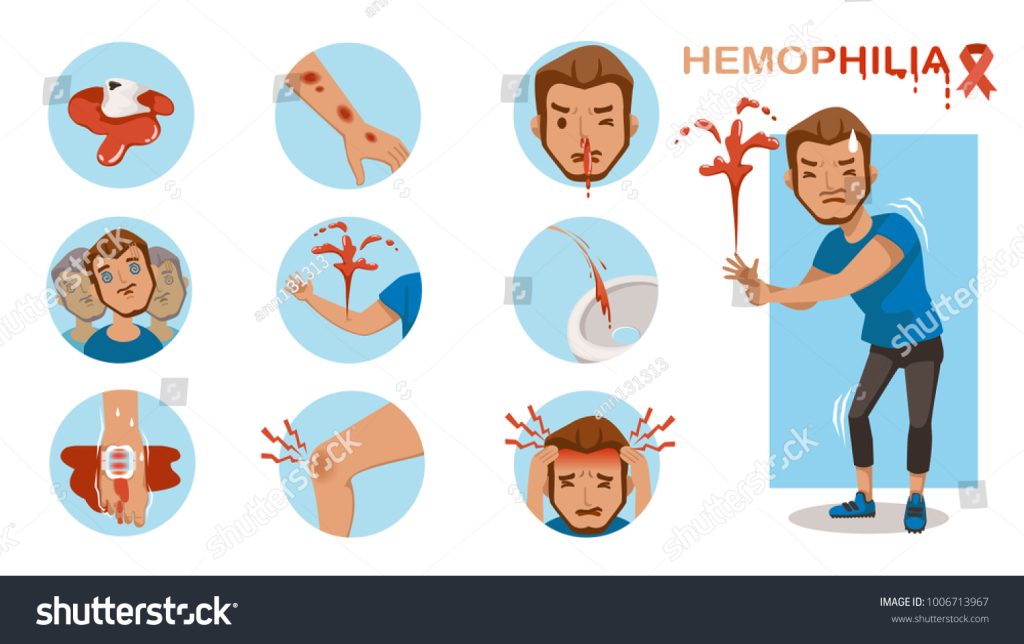
Individuals with hemophilia and their families can take several preventive measures to reduce the risk of bleeding episodes and manage the condition effectively:
a) Regular Medical Follow-up: Regular visits to a hematologist or a specialized hemophilia treatment center are crucial to monitor clotting factor levels and overall health.
b) Physical Activity: Engaging in low-impact exercises, such as swimming or biking, can help strengthen muscles and joints, reducing the risk of bleeding.
c) Injury Prevention: Taking precautions to avoid injuries and falls can significantly reduce the risk of bleeding episodes.
d) Medic Alert Identification: Wearing a medical alert bracelet or necklace can help inform medical professionals about the condition in case of emergencies.
Conclusion
Hemophilia is a complex bleeding disorder that requires proper diagnosis, management, and treatment to prevent complications and improve the quality of life for individuals affected by it. With advancements in medical science and research, the outlook for people with hemophilia continues to improve. Early diagnosis, appropriate treatment, and a supportive healthcare team are key to effectively managing this condition and allowing individuals with hemophilia to lead fulfilling lives.